# code from prior sectionsusingCSV, DataFrames, PhyloNetworks, PhyloPlots, RCallusingStatsBase, StatsModels, PhyloTraitsnet =readnewick("data/polemonium_network_calibrated.phy");for tip in net.node tip.name =replace(tip.name, r"_2$" => s""); endtraits = CSV.read("data/polemonium_traits_morph.csv", DataFrame)subset!(traits, :morph =>ByRow(in(tiplabels(net))))
For illustrative purposes, we are going to look at the evolution of mean elevation as a discrete variable. We discretize elevation using the threshold 2.3 km below, because the distribution of elevation shows somewhat of a gap around that value, and a reasonable number of morphs both below and above that value.
dat =DataFrame( morph =String.(traits[:, :morph]), elevation =map( x -> (x .>2.3 ? "hi":"lo"), traits.elevation ))
17×2 DataFrame
Row
morph
elevation
String
String
1
aff._viscosum_sp._nov.
hi
2
apachianum
hi
3
brandegeei
hi
4
carneum
lo
5
chartaceum
hi
6
confertum
hi
7
delicatum
hi
8
eddyense
hi
9
elegans
lo
10
elusum
lo
11
foliosissimum
hi
12
micranthum
lo
13
occidentale_occidentale
lo
14
pauciflorum
hi
15
pectinatum
lo
16
pulcherrimum_shastense
hi
17
reptans
lo
estimating the evolutionary rate(s)
For binary traits, we can use the :ERSM model, for “equal-rate substition model”:
fit =fitdiscrete(net, :ERSM, dat.morph, select(dat, :elevation))
PhyloTraits.StatisticalSubstitutionModel:
Equal Rates Substitution Model with k=2,
all rates equal to α=0.67674.
rate matrix Q:
hi lo
hi * 0.6767
lo 0.6767 *
on a network with 3 reticulations
data:
17 species
1 trait
log-likelihood: -11.05965
or the more general “binary trait substitution model” :BTSM that does not constrain the two rates to be equal:
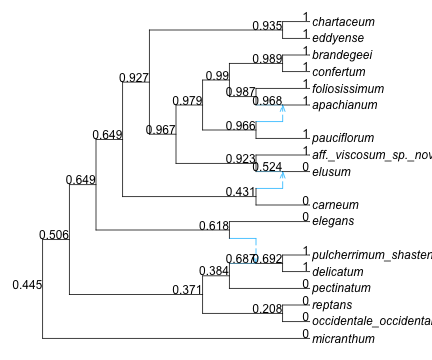
We can calculate ancestral state probabilities like this, but the output data frame is hard to interpret because it’s unclear which node has which number:
asr =ancestralreconstruction(fit)
39×4 DataFrame
14 rows omitted
Row
nodenumber
nodelabel
hi
lo
Int64
String
Float64
Float64
1
1
micranthum
0.0
1.0
2
2
occidentale_occidentale
0.0
1.0
3
3
reptans
0.0
1.0
4
4
pectinatum
0.0
1.0
5
5
delicatum
1.0
0.0
6
6
pulcherrimum_shastense
1.0
0.0
7
7
elegans
0.0
1.0
8
8
carneum
0.0
1.0
9
9
elusum
0.0
1.0
10
10
aff._viscosum_sp._nov.
1.0
0.0
11
11
pauciflorum
1.0
0.0
12
12
apachianum
1.0
0.0
13
13
foliosissimum
1.0
0.0
⋮
⋮
⋮
⋮
⋮
28
28
28
0.986652
0.0133477
29
29
29
0.96571
0.0342897
30
30
H18
0.968377
0.0316228
31
31
31
0.923395
0.0766053
32
32
32
0.431109
0.568891
33
33
H20
0.524334
0.475666
34
34
34
0.61782
0.38218
35
35
35
0.370785
0.629215
36
36
36
0.384265
0.615735
37
37
H19
0.686994
0.313006
38
38
38
0.691704
0.308296
39
39
39
0.208387
0.791613
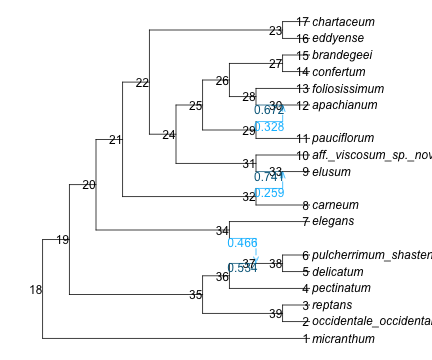
But we can map the posterior probability of state “hi” onto the network. It is important to use the network stored in the fitted object, to be assured that the node numbers match.
(b) showing the posterior probability of state ‘hi’
Figure 1: network stored in fitted object, to map ancestral state probabilities
effect of gene flow
What is the evidence that a trait was inherited via gene flow (along the minor hybrid edge) versus “vertically” (via the major hybrid edge)?
To answer this question, we can extract the prior probability of either option, and the posterior probabilities conditional on the data. Then, the Bayes factor to compare the 2 hypotheses, minor versus major edge (or gene flow versus vertical), can be expressed as an odds ratio: \[\mathrm{BF} = \frac{P(\mathrm{data} \mid \mathrm{minor})}{P(\mathrm{data} \mid \mathrm{mafor})} = \frac{\frac{P(\mathrm{minor} \mid \mathrm{data})}{P(\mathrm{major}\mid\mathrm{data})}}{\frac{P(\mathrm{minor})}{P(\mathrm{major})}}\,.\]
Here, let’s say we want to focus on the reticulation that is ancestral to elusum (node 33, H20). From Figure 1 (left) we see that the major (“vertical”) parent edge has γ = 0.741 = P(major) and the minor (“gene flow”) edge has γ = 0.259 = P(minor). So at this reticulation, the prior odds of the trait being inherited via gene flow is 0.259/0.741 = 0.35. Here how we may get this prior odds programmatically:
nodeH20 = net.node[findfirst(n->n.name=="H20", net.node)] # find node named H20priorminor =getparentedgeminor(nodeH20).gamma # γ of its minor parentpriorodds = priorminor / (1-priorminor)
0.34993311937527516
To extract the posterior odds and calculate the Bayes factor, we can do as follows, to focus on just 1 reticulation of interest and integrate out what happened at other reticulations: